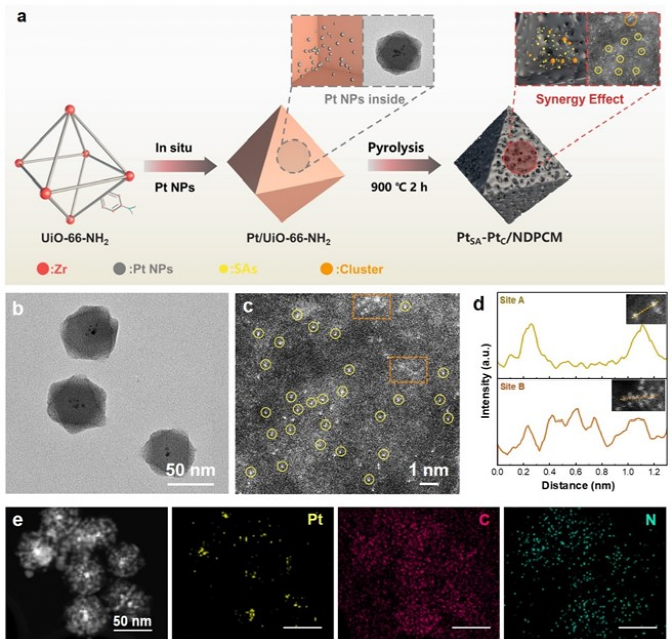
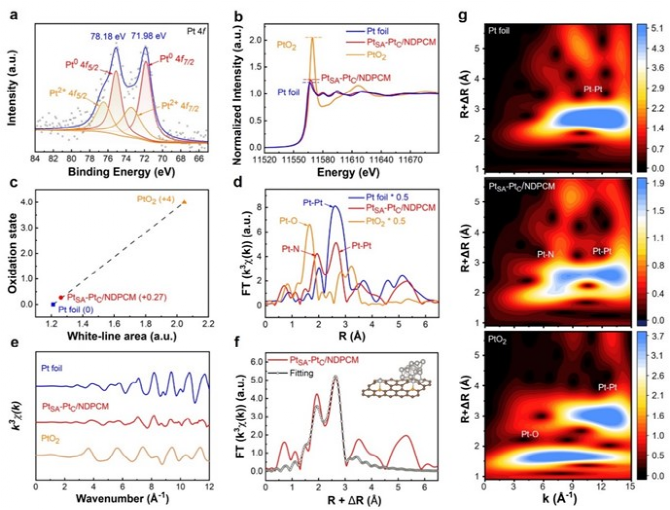
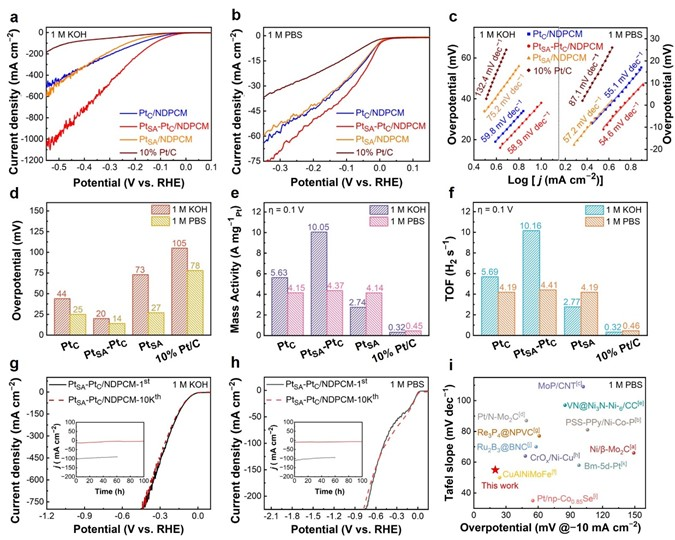
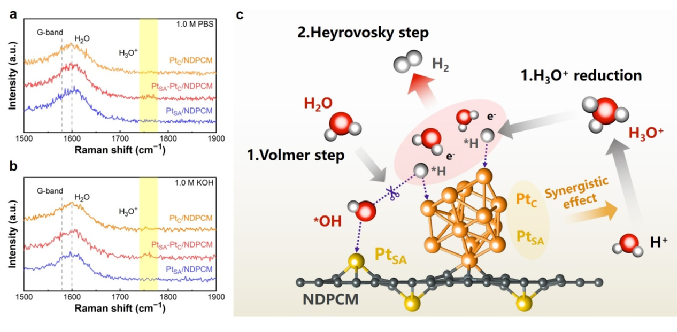
电催化析氢是获得化学燃料以实现能源可持续性和有效减少二氧化碳排放的一种很有前景的方案。在电催化析氢反应(HER)催化剂的各种金属中,铂(Pt)因其固有的超低H吸附吉布斯自由能(|ΔGH*|,~0.0071eV)而具有低过电位和快速动力学反应,是提高HER性能最有效的活性金属。然而,Pt金属的高成本、超低利用率和较差的循环稳定性严重限制了其进一步大规模应用。单原子Pt (PtSA)可以最大限度地提高原子利用效率,从而降低催化剂成本并保持其优越的HER活性。目前,诸多研究主要集中在通过抑制合成过程或电催化反应过程中单原子(SA)的团聚来提高SAC的电催化活性,其中能将SA限制在载体内部而提高SAC的循环稳定性则显得尤为重要。金属有机框架(MOFs)具有周期性的孔隙和独特的结构,被认为是SA实现循环稳定性的理想载体,而且根据不同种类配位金属的选择,可以设计成各种功能性杂化配合物。此外,MOFs也被认为是封装纳米粒子(NPs)的新一代理想载体,与传统的多孔材料NPs与MOFs的相互作用也可以促进质子和电荷的转移。 近日,威尼斯886699廉孜超课题组等人通过将Pt NPs原位封装进UiO-66-NH2后进行高温烧结,构建了具有Pt单原子和团簇封装在N掺杂的多孔碳催化剂。通过将Pt单原子和团簇封装在N掺杂的多孔碳核心中,有效提高了材料在碱性与中性条件下的电催化析氢性能和循环耐久性。其中达到电流密度为-10 mA cm-2时, PtSA-PtC/NDPCM 在碱性条件下表现出20 mV的超低过电势,且在中性条件下过电位仅为14 mV。此外,该催化剂在电流密度-10 mA cm-2 时,1.0 M KOH和1.0 M PBS中分别进行100小时或10,000次CV循环后,性能无任何明显衰减。相对于最近报道的电催化析氢催化剂, PtSA-PtC/NDPCM具有出色的优势。DFT结果表明,Pt单原子(PtSA)和Pt团簇(PtC)的共存,可以提供多个H吸附位点,并调节活性位点的电子状态,从而有利于形成极低的ΔGH*。原位拉曼光谱进一步表明,PtSA和PtC的协同作用创造了局部的酸性微环境,使其在碱性或中性电解质中引发了H3O+诱导的水还原反应,加速了HER反应过程。该文章发表在国际顶级期刊Adv. Funct. Mater. 上,杨伟伟,李梦媛,张毕堃,刘亚子,訾江芝为本文共同第一作者。 图1(a)所示为Pt单原子和团簇封装在N掺杂的多孔碳催化剂的合成策略。其中所合成的Pt@UiO-66-NH2粒径大小为61.6 nm(图1(b))。图1(c)中PtSA-PtC/NDPCM的高角度环形暗场扫描透射电子显微镜(HAADF-STEM)图像显示, PtSA和PtC均匀地分散在NDPCM中,分别用黄色圆圈和橙色矩形标记。此外,PtSA的强度峰具有相似的高度和较大的距离,而PtC的强度峰由于轻微聚集而显示出不同的高度和较小的距离(图1(d)),进一步确认PtSA和PtC的形成。能量色散X射线(EDX)元素图谱表明,铂元素主要均匀地分散在NDPCM的核心中(图1(e))。图1 (a) PtSA-PtC/NDPCM的合成程序示意图;(b) Pt@UiO-66-NH2的TEM图;(c) NDPCM中PtSA和PtC的HAADF-STEM图像。单分散的PtSA用黄色的圆圈表示,而PtC则用橙色的矩形标记;(d) 由(c)的插图HAADF-STEM图像得到的强度曲线;(e) 相应元素的EDX元素图谱。 如图2(a)所示,高分辨率的Pt 4f光谱中,71.98和75.18 eV的两个峰对应着Pt0,而73.38和76.38 eV的峰值则归因于Pt2+的存在,表明PtSA-PtC/NDPCM中共同存在着金属铂和离子铂。图2(b)所示,X射线吸收近边结构(XANES)的白线峰强度可以揭示Pt未占用的5d轨道的密度,其中PtSA-PtC/NDPCM的Pt L3边WL强度更接近于Pt箔,但低于PtO2,表明材料中有大量未被占据的5d电子态。经过计算,PtSA-PtC/NDPCM中Pt的氧化态约为+0.27(图2(c))。此外,通过Pt的扩展X射线吸收精细结构(EXAFS)光谱显示, R=1.94 Å的峰值对应于Pt-N键,而2.65 Å的峰值对应Pt-Pt键(图2(d)),证实了Pt单原子和Pt团簇同时存在于PtSA-PtC/NDPCM。EXAFS的拟合结果表明中心铂原子的第一壳层配位数为16,包括1个N原子和15个铂原子(图2(f))。结合HAADF-STEM和XAS的表征结果,PtSA-PtC/NDPCM中PtSA和PtC的存在可以得到充分的证明。图2 (a) PtSA-PtC/NDPCM的Pt 4f轨道的高分辨率XPS光谱;(b) Pt箔、PtSA-PtC/NDPCM和PtO2的归一化Pt L3边的XANES光谱;(c) 从PtSA-PtC/NDPCM的ΔXANES光谱以及作为参考的Pt foil和PtO2的光谱计算出的Pt氧化态;(d) Pt箔、PtSA-PtC/NDPCM和PtO2中相应的k3-weight FT-EXAFS,没有相位校正;(e) PtSA-PtC/NDPCM在Pt L3边的K空间和(f) R空间的EXAFS拟合曲线;(g) Pt箔、PtSA-PtC/NDPCM和PtO2的小波变换图。 PtSA-PtC/NDPCM在1.0 M KOH中对HER表现出出色的活性,在-10 mA cm-2时过电位为20 mV,在~540 mV时电流密度可以达到-913 mA cm-2(图3(a)和图3(d))。此外,令人惊讶的是PtSA-PtC/NDPCM在1.0 M PBS中也显示出超低的过电位,在-10 mA cm-2时仅为14 mV(图3(b)和图3(d))。根据Volmer、Heyrovsky和Tafel步骤的Tafel斜率的理论值,PtSA-PtC/NDPCM在碱性或中性条件下反应机理为更有利的Volmer-Heyrovsky步骤。此外,如图3(e)和图3(f)所示,在η = 100 mV时PtSA-PtC/NDPCM显示出最高的质量活性和周转频率(TOF)。图3 PtC/NDPCM、PtSA-PtC/NDPCM、PtSA/NDPCM和10% Pt/C在 (a) 1.0 M KOH和(b) 1.0 M PBS中的极化曲线;(c) 从相应的LSV曲线得到的PtC/NDPCM、PtSA-PtC/NDPCM、PtSA/NDPCM和10% Pt/C的Tafel图;(d) 各种催化剂在1.0 M KOH和1.0 M PBS中达到-10 mA cm-2所需的过电位;(e) 在1.0 M KOH和1.0 M PBS中,η = 100 mV时,催化剂的质量活性;(f) 在1.0 M KOH和1.0 M PBS中η = 100 mV的TOF图;(g) PtSA-PtC/NDPCM在1.0 M KOH中以10 mV s-1的扫描速率在-0.1和-0.5 V之间进行10,000次CV扫描之前和之后的极化曲线。插入图:PtSA-PtC/NDPCM在j = -10和-100 mA cm-2时分别进行的i-t测试;(h) PtSA-PtC/NDPCM在1.0 M PBS中以10 mV s-1的扫描速率在-0.1和-0.5 V之间进行10,000次CV扫描之前和之后的极化曲线。插入图:PtSA-PtC/NDPCM分别在j = -10和-100mA cm-2时的i-t测试;(i) PtSA-PtC/NDPCM在1.0 M PBS中的HER活性与其他催化剂的比较。 拉曼光谱中,在1.0 M KOH或1.0 M PBS中三种不同的电位条件下,可以清楚地观察到NDPCM的G带(1580 cm-1)和H2O的峰(1600 cm-1),而施加的电位逐渐降低时,可检测到中间物种H3O+的峰(1750 cm-1),且NPDCM的G带变得更宽,表明催化剂产生大量异常的H3O+中间物(图4(a)和图4(b))。此外,PtC/NDPCM和PtSA/NDPCM在-30 mA cm-2处没有检测到明显的H3O+峰,证明PtSA-PtC/NDPCM中单原子和团簇的协同作用在HER过程于中性或碱性条件下构建了局部酸性微环境。 结合Tafel斜率值和DFT计算结果,我们推测详细的Volmer-Heyrovsky反应过程如下(图4(c))。水分子最初被吸附在单原子位点上,根据Volmer步骤,解离的H*和OH*分别被吸附在团簇和单原子位点上。由于PtSA-PtC/NDPCM的强水解能力,产生了大量的H*,导致一些应该吸附H*的团簇位点很快被Hupd覆盖。因此,H+只能与附近的水分子结合,且单原子和团簇的协同作用诱发了大量的H3O+,在中性或碱性条件下形成了局部的酸性微环境。随后,由于电子被转移到表面,H3O+被还原成H*,而H*则被吸附在团簇的位置。最后,根据Heyrovsky步骤,H*然后与另一个水分子和一个电子反应,形成H2。鉴于PtSA-PtC/NDPCM中存在多个活性位点以及PtSA和PtC的协同作用,水的吸附/解吸与H3O+诱导的水在局部酸性微环境中的还原同时发生,有效地提高了HER在碱性和中性条件下的催化活性。图4 PtC/NDPCM、PtSA-PtC/NDPCM、PtSA/NDPCM在-30 mA cm-2下在(a) 1.0 M PBS和(b) 1.0 M KOH中的拉曼光谱;(c) PtSA-PtC/NDPCM的HER过程的机制示意图。 结果表明,PtSA-PtC/NDPCM在HER中表现出极高的电催化活性,在碱性和中性电解质下具有低过电位和长期稳定性。实验结果和DFT计算表明,PtSA-PtC/NDPCM中的PtSA和PtC分别适合在不同的pH条件下析氢,且两者的协同作用在中性和碱性条件下构建了一个局部的酸性微环境,有利于同时发生水的吸附/解吸和H3O+诱导的水还原。这项研究为显著提高制氢效率铺平了道路,为开发新型的HER电催化剂提供了灵感。 Weiwei Yang, Mengyuan Li, Bikun Zhang, Yazi Liu, Jiangzhi Zi, Han Xiao, Xinyang Liu, Jingkai Lin, Huayang Zhang, Jian Chen, Zhengfen Wan, Zhen Li, Guisheng Li, Hexing Li, Zichao Lian, Interfacial Microenvironment Modulation Boosts Efficient Hydrogen Evolution Reaction in Neutral and Alkaline, Adv. Funct. Mater., 2023. https://doi.org/10.1002/adfm.202304852 李贵生 博士生导师,威尼斯886699威尼斯886699副院长(主持工作),上海师范大学环境与地理科学学院兼职教授,主要从事环境催化研究工作,基于光/光电环境催化绿色技术,对环境污染物(水和气)进行控制处理与资源化,实现有机污染物深度矿化与分解水制氢、CO2还原制备燃料等的协同研究。以通讯或第一作者身份在环境与能源各类重要国际SCI期刊上发表了近70余篇高质量论文,其中有11篇论文入选全球引用前1%ESI高被引论文,1篇论文1‰ ESI热点论文,被同行他引用达6000余次,h指数46。目前完成及主持国家自然科学基金青年、面上项目共计4项,获得中国专利授权20项。 李和兴 国家杰出青年基金获得者,现任上海电力大学校长。担任Appl. Catal. B副主编、教育部化学化工学部委员、国家“111引智基地”负责人、教育部重点实验室和国际联合实验室主任、中国光催化专委会副主任、上海市电力科协主席。从事光催化污染治理和化学污染源头控制研究,承担国家自然科学基金重点项目和国际合作重点项目、科技部重大研发计划课题;发表SCI论文200余篇,包括Nat. Sustain.、Nat. Commun.、Angew. Chem. Int. Ed.、J. Am. Chem. Soc.、Adv. Mater.、Adv. Funct. Mater.等,他引19000余次,H因子73,入选Elsevier化工领域中国高被引用作者;主编英文专著2部、中文专著5部,授权发明专利63件,其中转让9件。获教育部自然科学一等奖及上海市自然科学一等奖等。 廉孜超 威尼斯886699威尼斯886699化学系特聘教授,上海市QR特聘专家、上海市东方学者特聘教授、浦江人才和日本JSPS特聘研究员,PI。主要研究领域:合成无机化学和超快光谱学研究:设计光催化剂和研究光生载流子动力学应用于光催化或光电催化水分解制氢气和环境催化。至今共发表SCI论文34篇,以一作或通讯发表SCI论文21篇,包括Nature Sustain.、Nature Commun.、Commun. Chem.、JACS(2篇)、ACS Nano、Adv. Funct. Mater.(4篇)、Appl. Catal. B-Environ.(3篇)等,IF>10的15篇,论文他引1435次,3篇曾为ESI论文,获JACS、JPCC等封面文章,荣获上海市自然科学二等奖(3/4),主持国家自然科学基金项目1项、上海市人才项目2项和上海市基金2项等,获授权专利6件。JACS、Angew等特邀审稿人。 https://www.x-mol.com/groups/Lian_Zichao 杨伟伟:博士,威尼斯886699讲师,硕士生导师。研究方向为能源的转化与存储,具体包括电催化析氢、锂硫电池和锂离子电池。至今以第一作者在Nature Commun.、Adv. Mater.、Adv. Energy Mater.、Adv. Funct. Mater.、J. Mater. Chem. A、Chem. Eng. J. 等国际知名期刊上发表学术论文多篇,他引300多次。主持国家自然科学基金青年基金1项,上海市自然基金面上项目1项,申请国家发明专利4项,授权2项。发表成果被科学网和能源学人等学术媒体多次转载报道。 李梦媛:威尼斯886699廉孜超教授课题组硕士研究生,研究方向为纳米功能材料合成和电解水析氢,以第一/共一作者在Adv. Funct. Mater等国际知名期刊上发表学术论文多篇。
来源:能源学人
链接:李和兴/李贵生/廉孜超Adv. Funct. Mater.:界面微环境的调控以促进中性和碱性环境下的高效析氢